What is hereditary colorectal cancer?
All of the body’s cells normally grow, divide, and then die in order to keep the body healthy and functioning properly. Sometimes this process gets out of control: cells keep growing and dividing even when they are supposed to die. When the cells lining of the colon multiply uncontrolled, cancer can ultimately develop in any part of the colon [Figure 1]. Colorectal cancer ranks as the second to third most common cancer in the world depending on the geographic location. It affects both men and women. In the USA approximately 1 in 20 people will develop colorectal cancer.
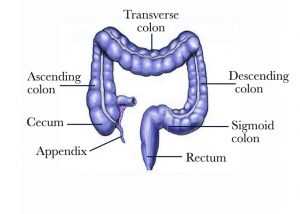
Figure 1 – Picture of the large bowel
The majority of patients with colorectal cancer have no family history of colorectal cancer. Such patients are classified as sporadic colorectal cancer and typically present with cancer at age 50 or older. Some patients have a family history of colorectal cancer in 1 or more relative. This history is significant especially if the cancer occurred before age 50. A minority of patients (5 to 10%) have hereditary colorectal cancers with are associated with a specific inherited genetic abnormality. The abnormal gene is inherited from 1 or both parents and leads to the development of colorectal cancer. In addition, patients with inherited colorectal cancer syndromes are at increased risk of developing cancer in other organs.
Our understanding of hereditary colorectal cancers is evolving and our scientific knowledge is growing as new genes are identified. Currently several genetic syndromes have been identified and they include the following:
Polyposis Syndromes (abnormal gene that causes polyps and cancer)
- Attenuated familial adenomatous polyposis
- Familial adenomatous polyposis
- Juvenile polyposis syndrome
- MYH-associated polyposis
- Peutz-Jeghers syndrome
- PTEN Hamartoma Tumor Syndrome, Cowden Syndrome, and Bannayan-Riley-Ruvalcaba Syndrom
- Serrated polyposis syndrome/hyperplastic polyposis syndrome
Nonpolyposis Syndromes (cancer with or without polyps)
- Familial colorectal cancer
- Familial colorectal cancer type X
- Hereditary Nonpolyposis Colorectal (Lynch syndrome)
Below is a review of a few of these syndromes. If after reading the below sections, you need more extensive resources on the topic, you can check https://www.cancer.gov/types/colorectal/hp/colorectal-genetics-pdq#section/all?redirect=true
Why is the medical history very important?
The first step in determining whether a patient has predisposition to developing colorectal cancer is to review the family history. While some patients may be the first family member to develop colorectal cancer, a detailed family history is a very critical step. It is important to gather information on each family member with specific attention to the type of cancer, location, and age at diagnosis. A list of all family members with colorectal polyps or cancer, breast, ovarian, or uterine cancer should be reviewed. A significant family history that raises suspicion for a hereditary colorectal cancer syndrome is when multiple members in more than 1 generation are affected by colorectal cancers, polyps, or 1 or more of the listed cancers. A genetic counselor is the professional who can guide a patient through this process and determine whether the history is significant enough to recommend genetic testing. Before and after genetic testing, proper counseling is needed in order for the patient to appreciate the ramifications of such testing to the patient, his/her children and other family members.
Attenuated Familial Adenomatous Polyposis (aFAP)
Patients with the attenuated form of familial adenomatous polyposis (aFAP) usually present with an average of 20 to 30 polyps (a range between 10 and 100). In many cases, this syndrome is due to a mutation in the APC gene which functions to control cell growth. It is inherited in an autosomal dominant fashion which means 1 abnormal gene in 1 parent is sufficient to cause the disease if passed from parent to child. But compared to FAP (see section on Familial Polyposis Syndrome), fewer patients with aFAP test positive for this mutation. Some patients with aFAP are the first family member to have the abnormal APC gene. Others may test positive for the MYH mutation (see MYH Associated Polyposis or MAP). The polyps in aFAP are more often found in the right side of the colon and patients with aFAP tend to present between the age of 35 and 45 years. Colorectal cancer typically develops in the 6th decade of life. Due to a right sided predominance, Colonoscopy is advisable starting between the age of 20 and 30, or 10 years sooner than the first polyp or cancer diagnosis in a relative. The frequency of follow-up colonoscopy varies from 1 to 3 years. aFAP patients do frequently have upper intestinal polyps (in the stomach and the first portion of the small intestine called the duodenum), Gastroscopy is recommended and should be repeated every 2 to 3 years. Colonic polyps in aFAP may be manageable with removal during colonoscopy and removal of the colon and/or rectum is reserved for patients whose condition cannot be controlled endoscopically (large lesions, numerous recurrences) or those with cancer diagnosis.
For additional information on this condition, https://rarediseases.info.nih.gov/diseases/8532/attenuated-familial-adenomatous-polyposis
Familial Adenomatous Polyposis (FAP)
FAP is an inherited condition. Polyps develop in the gastrointestinal tract both the stomach and the small intestine (predominantly the duodenum) and the large bowel (colon and rectum). The large bowel is primarily affected with at least 100 but often hundreds or greater than 1000 polyps. FAP is caused by a genetic mutation in the APC gene, which controls the rate of cell growth [Figure 2]. It is an autosomal dominant disease. Inheriting one abnormal gene from one parent is sufficient to cause this condition. APC testing is positive in 80% of patients with > 1,000 polyps and <60% of patients with 100 to 1,000 polyps. It is important to note that in 20-30% of the patients develop this gene mutation during conception and both parents are normal. Adenomatous precancerous polyps grow in the lining of the colon and rectum. Untreated the majority of patients develop colorectal cancer by age 40, although many patients develop cancer in their 20’s or 30’s. Since FAP runs in families, it is important to have other relatives tested. Early detection and treatment can prevent progression to cancer. Genetic blood testing and endoscopic screening starting in puberty is critical as young patients have no symptoms initially. Potential symptoms include diarrhea, altered bowel habits, mucous discharge, abdominal pain and cramps, anemia, and bloody bowel movements. Initial screening for FAP include Flexible Sigmoidoscopy or Colonoscopy which is done yearly in patients with a documented gene mutation. Patients with FAP should undergo Gastroscopy to check for stomach and duodenal polyps which is advisable starting age 20. Based on the initial findings, this examination should be repeated every 1 to 2 years. Patients with documented FAP benefit from surgical removal of the large bowel. Minimally Invasive Laparoscopic Surgery (keyhole camera surgery) is Dr. Maher Abbas’ preferred surgical approach as it yields faster recovery and less complications. Surgical options include removal of the colon and connecting the small bowel to the rectum, or removal of the colon and the rectum and connecting the small intestine to the anus after fashioning a new rectum from the small intestine (ileal pouch). The timing of surgery depends on how extensive the polyps, the age of the patient, and the presence or concern for cancer in some polyps.
Although FAP always affects the colon, the gene mutation is present in the entire body. Other organs can be potentially affected including the liver and pancreas, brain, thyroid gland, adrenal gland, bones, skin, and teeth. Congenital hypertrophy of the retina (a flat pigmented spot in the outer layer of the retina) is present in some patients. Desmoid tumors (fibrous or scar-tissue-like tumors) are rare tumors that occur in 15-30% of FAP patients and predominantly involve the abdomen. They can lead to bowel blockage or fistula. The treatment of desmoid tumors is complex and it involves a combination of medical therapy and in some cases surgery.
For additional information on this condition, https://rarediseases.info.nih.gov/diseases/6408/familial-adenomatous-polyposis

Figure 2 – Colonoscopy view of a patient with familial polyposis syndrome and numerous polyps
Juvenile Polyposis Syndrome (JPS)
Patients with JPS develop polyps in the gastrointestinal tract, anywhere from the stomach to the rectum. The word “juvenile” refers to the way the polyps look under the microscope and not to the age of the patient (although most patients with JPS develop symptoms by 20 years of age). The most common location for these polyps is the large bowel, the stomach, and less commonly the small intestine. The polyps vary in shape and size, can be flat or on a stalk, and can range from a few to more than a hundred.
JPS is caused by a defective gene which disrupt cell growth and death. Normal tissue grows in an uncontrolled way and lead to polyp formation. About 75% of people with JPS have a family history of the disorder. If 1 of the parents has a gene defect, the children have 50% chance of inheriting JPS. About 25% of the time, the gene does not get inherited but occurs at the time of formation of the baby. The 2 genes that have been linked to JPS are BMPR1A and SMAD4. The presenting symptoms of JPS include blood in the stool, cramping abdominal pain, constipation, diarrhea, fatigue, and weight loss. Findings include anemia (low blood count) and in some cases prolapsing polyps are noted through the anus.
JPS is diagnosed by undergoing Colonoscopy and Gastroscopy. These 2 procedures carefully examine the upper gastrointestinal tract including stomach and duodenum and the lower gastrointestinal tract including the colon and rectum. The first endoscopic examination should be at age 15. The diagnosis of JPS is made in a patient who has 5 or more juvenile polyps in the colon and rectum, multiple polyps in the upper and lower gastrointestinal tract, or has any number of juvenile polyps in the setting of family history of JPS. Periodic endoscopic examinations are recommended to remove the polyps. Patients with extensive polyposis require surgical excision. It is important to note that there is a risk of 10-50% of developing cancer in the polyps. Surveillance with endoscopy is performed between 1 and 3 years depending on the findings.
In patients with SMAD4 mutations, additional recommended studies are MRI of the brain to check for vascular malformations at the time of JPS diagnosis (if normal, no need to repeat), echocardiography (an ultrasound examination of the heart) to check for vascular malformations of the lung, ultrasound of the liver and annual CBC (complete blood count) in patients over age 35 to check for any evidence of bleeding from gastrointestinal arteriovenous malformations.
For additional information on this condition, https://rarediseases.info.nih.gov/diseases/3065/juvenile-polyposis-syndrome
Hereditary Nonpolyposis Colorectal Cancer (Lynch syndrome)
Hereditary Nonpolyposis Colorectal (HNPCC referred to as Lynch syndrome) is an inherited condition that increases the risk of cancer of certain organs. A genetic mutation in 1 of the genes that fixes DNA duplication errors causes this syndrome. Accumulation of damaged cells lead to cancer. A genetic mutation in the mismatch repair (MMR) gene is present in patients with the Lynch syndrome. The specific genes associated with Lynch syndrome are MLH1, MSH2, MSH6, PMS2, and EPCAM. Lynch syndrome is an autosomal dominant disorder. This means that if only 1 parent carries an abnormal gene, there’s a 50 percent chance will inherit the mutation.
Patients with Lynch syndrome may have adenomatous polyps in the colon or rectum. The right side of the colon is the most common location for polyps or cancer. Polyp formation occurs faster than in average risk patients (1-2 years versus 10 years) and patients who have 1 colorectal cancer have an increased risk of developing another cancer after the initial surgery (15 percent within 10 years, 40 percent within 20 years, and 60 percent after 30 years). This risk depends on the type of the first surgery performed.
HNPCC is defined by the occurrence of multiple cancer cases in the family that meets the following criteria:
- > 3 relatives have colorectal or other HNPCC-related cancers such as endometrial cancer and 1 patient being a first-degree relative (parent, child, or sibling) of the other 2
- > 2 generations are affected
- > 1 person with colorectal cancer before age 50
- Familial adenomatous polyposis has been excluded
Patients are at risk of certain cancers. The lifetime risks and age at presentation depend on the type of MMR mutation:
- Colorectal cancer. From 10 to 74% of patients will be affected with an average age at diagnosis between 27 and 66 years. Colorectal cancer risk in the general population is 5%
- Endometrial cancer. From 14 to 71% of women will be affected with an average age at diagnosis between 48 and 62 years. Endometrial cancer in women in the general population is 2.7%
- Ovarian cancer. From 4 to 20% of women will be affected with an average age at diagnosis between 43 and 45 years. Ovarian cancer in women in the general population is 1.5%
Less common cancers include stomach (0.2-13% with average age of diagnosis 49 to 55 years), small bowel (0.4-12% with average age of 46 to 49 years), hepatobiliary (0.02-4% with average age of 54 to 57 years), pancreas (0.4-4% with average age 63 to 65 years), urinary tract (0.2-25% with average age of 52 to 60 years), brain and central nervous system (1-4% with average age of 50 years), skin cancer (1-9%), prostate (9-30% with average age of 59 to 60 years), breast (5-18% with average age of 52 years). It is unclear if prostate and breast cancer are associated with this genetic abnormality. Some individuals with Lynch syndrome develop skin growths such as sebaceous adenomas, keratoacanthomas, and skin cancers like sebaceous carcinoma. Such patients are classified as Muir-Torre syndrome.
Screening tests and genetic testing are available to diagnose Lynch syndrome. A piece of the tumor is tested for the defective gene responsible for DNA repair looking for changes called microsatellite instability (MSI). Tumors with microsatellite instability are called MSI-high (MSI-H). 90 to 95% of colorectal cancers of Lynch syndrome patients are MSI-H. However, since 5 to 10 percent of Lynch syndrome tumors do not show instability, a negative MSI-H test cannot completely rule out the possibility. Immunohistochemistry (IHC) testing is a screening test that checks for proteins that are missing in patients with Lynch syndrome. Approximately 88 percent of individuals with Lynch syndrome will have an abnormal IHC result. In addition to testing abnormal tissue removed from the patient (a polyp or a tumor), genetic testing and counseling can be conducted. A blood test or a brushing from the inside of the mouth can determine if a MLH1, MSH2, MSH6, PMS2, or EPCAM gene mutation is present in the family.
Patients with Lynch syndrome should undergo their 1st colonoscopy at age 20 and repeat every 1-2 years until the age 40. Yearly colonoscopy after age 40. Women should have yearly transvaginal ultrasound and a CA-125 blood test beginning at age 30. Furthermore, women with Lynch syndrome should consider removal of the uterus and the ovaries after age 35 years or once childbearing is complete. Other recommendations include gastroscopy every 2-3 years starting at age 30 and capsule endoscopy to look at the small intestine every 2-3 years, yearly skin exam, and urinalysis every year beginning at age 35.
For additional information on this condition, https://rarediseases.info.nih.gov/diseases/9905/lynch-syndrome
MYH Associated Polyposis (MAP)
MAP is a rare hereditary condition which is related to a mutation in the MYH gene. The MYH gene is responsible for the repair of oxidative damage to the DNA. Mutations in this gene prevent cells from correcting mistakes that are made when DNA is copied (DNA replication) in preparation for cell division. As these mistakes build up in a person’s DNA, the likelihood of cell overgrowth increases, leading to colon polyps and the possibility of colon cancer. Patients with this gene mutation develop various types of polyps in the large bowel including hyperplastic polyps, sessile (flat) serrated polyps, and adenomas. Most individuals with MAP develop between 10 and 100 polyps, although some can form over 1,000 polyps. Age of diagnosis is usually in the late 40’s. Although MAP patients can have findings similar to FAP, the MAP gene is inherited in a recessive manner and both parents need to pass the gene for a child to develop the syndrome. MAP increases the risk of developing colorectal cancer between the 5th and 7th decade of life. An estimated 50% of people with MAP will have colorectal cancer at the time of diagnosis. Patients with MAP are at increased risks of developing polyps in the stomach and the duodenum. While the stomach fundic gland polyps are usually of little significance, the duodenal polyps are precancerous and undetected they can lead to cancer. Other affected organs are the thyroid gland, and the kidneys. MAP can be confirmed by genetic testing (blood test or brushing from inside of the mouth) and should be suspected in patients who have more than 10 adenomatous polyps and negative APC gene testing. MAP is diagnosed when a person is found to have two MYH gene mutations. A carrier (a person with 1 defective gene) is not believed to have an increased risk of colorectal cancer or polyps. Individuals with MAP are best monitored with colonoscopy starting at age 20 or 10 years prior the youngest diagnosis in the family, whichever is earlier. It is important that people with MAP have lifelong colonoscopies because new polyps can form that may turn into a cancer. If less than 20 polyps are found, they can be removed individually during a colonoscopy. If the polyps are too numerous or too fast growing, then surgical removal of the colon and/or rectum might be necessary. The timing and choice of colon surgery depend on several factors, primarily the number and size of the polyps. Laparoscopic surgery has made removal of the colon a lot less painful and less disabling. Even after surgery the remaining bowel is checked yearly. Gastroscopy is recommended beginning at age 20 and every 1-3 years, depending on the size, number and microscopic analysis of the duodenal polyps. All MAP patients should have a baseline thyroid ultrasound starting at the age of 20. This should be repeated yearly, or more frequently if abnormalities are detected.
For additional information on this condition, https://rarediseases.info.nih.gov/diseases/10805/myh-associated-polyposis, https://www.cancer.net/cancer-types/mutyh-or-myh-associated-polyposis
Peutz-Jeghers Syndrome (PJS)
Peutz-Jeghers syndrome (PJS) is a condition characterized by gastrointestinal polyps (stomach, small bowel, colon), dark-colored spots, and certain cancers. Polyps can involve the lungs, nose, and bladder. The polyps are of the hamartomatous type which is a benign growth. 50 percent of individuals with PJS have a parent with the syndrome. The other 50 percent of people diagnosed with PJS are the first in the family with the syndrome. There is a 50 percent chance that a child of someone with PJS might inherit the mutated copy of the gene. A mutation in the STK11/LKB1 gene is responsible for JPS.
Patients with JPS have dark-colored spots (brown or blue, called muco-cutaneous pigmentation) on various parts of the body. The spots can fade in the late teenage years. Involved parts of the body include the mouth, lips, eyes, nose, hands and feet, and anus. The polyps in the gastrointestinal tract can cause small bowel obstruction, small bowel intussusception (a condition where the small bowel turns itself inside out), bleeding, and anemia. The diagnosis of PJS is considered with > 2 hamartomatous polyps are present, a family history of PJS, and the presence of dark-colorectal spots. Genetic testing is done through a blood sample. Nearly all patients with PJS will have a change (mutation) in the STK11/LKB1 gene. Patients with JPS should undergo colonoscopy and gastroscopy for polyp removal. Polyps in the small intestine can be removed surgically. Since these polyps are benign, removal of the polyps without bowel resection can be considered. Screening with colonoscopy and gastroscopy starts in late teenage years and is repeated every 2 to 3 years. Capsule endoscopy or MRI enterography can begin by age 10 and repeated every 2 to 3 years. Additional recommendations include breast exams in women twice a year beginning at age 25 years with annual mammogram and breast MRI beginning at age 25 years and annual pelvic exams with annual transvaginal ultrasound beginning between the age of 18 and 20 years. Males should start annual testicular examination beginning at age 10 years. It is important to note that patients with JPS have an increased risk of developing cancer of the digestive tract and other organs. The lifetime risk of developing any sort of cancer is estimated to be as high as 93 percent. PJS patients have a 39% risk of colon cancer, a 36 percent risk of pancreatic cancer, and a 29% risk of developing stomach cancer. Women have as high as a 54 percent lifetime risk of developing breast cancer. Men and women have an increased risk of developing lung cancer, even without smoking.
For additional information on this condition, https://rarediseases.info.nih.gov/diseases/7378/peutz-jeghers-syndrome
Serrated Polyposis Syndrome (Hyperplastic Polyposis Syndrome)
Serrated polyposis syndrome (SPS), previously known as hyperplastic polyposis syndrome is a rare condition that is characterized by serrated colorectal polyps. Serrated polyps are defined by their saw-toothed appearance under the microscope. The various types of serrated polyps include hyperplastic polyps, commonly located on the left side of the colon, sessile serrated polyps [SSPs] (larger polyps on the right side of the colon), and serrated adenomas (traditional serrated adenomas or TSAs). Serrated lesions can be difficult to find on endoscopic examination as they are flat but can progress to cancer quickly. A patient meeting any one of the following criteria is diagnosed with SPS:
- > 20 serrated polyps located anywhere in the colon
- > 5 serrated polyps proximal to the sigmoid colon, at least 2 > 1 cm in size
- Any serrated polyp in the colon in a patient with a family history of SPS
Patients with SPS have a higher risk of developing colorectal cancer and should have a colonoscopy every year once a diagnosis is made. It might be necessary to have colonoscopies every 3 to 6 months initially. Age of diagnosis is usually in 6th or 7th decade of life. Various studies have suggested that SPS can be inherited. First-degree relatives (i.e., parents, siblings and children) of someone with SPS have an increased risk of developing SPS or polyps. Approximately 40% of first-degree relatives of people with SPS had polyps found on screening colonoscopy. Colonoscopy examination should start at age 40 when a family history is present or 10 years earlier than a diagnosis of colorectal cancer in an SPS family member. Following a baseline exam, colonoscopy should be repeated every five years if no polyps are found. If serrated polyps or multiple adenomatous polyps are found, consider colonoscopy every 1 to 3 years. SPS patients have an increased risk of developing colorectal cancer. About 25-40% of people with SPS will develop colorectal cancer. There are not any other cancer risks that have definitively been established to be associated with SPS and screening of other organs is not recommended.
Why seek care with a colorectal surgeon?
Colorectal surgery is the specialty that deals with hereditary colorectal cancer. Colon and rectal surgeons are experts in the surgical and non-surgical treatment of diseases of the colon, rectum, and anus. Dr. Maher Abbas is an American Board Certified Colon and Rectal Surgeon with special interest in colorectal cancer and polyps. He provides the entire spectrum of care including colonoscopy and surgery. Although Dr. Maher Abbas does not provide genetic counseling or testing, he routinely provides care to young patients with extensive polyposis or early onset colorectal cancers. In addition to providing endoscopic services for screening and surveillance, Dr. Maher Abbas offers the entire spectrum of surgical interventions when needed. In addition, patients are counseled regarding chemoprevention which is the use of certain non-steroidal anti-inflammatory medications such as sulindac, aspirin, and curcumin to help reduce the formation of adenomatous polyps in some patients.

If you would like to schedule a consultation with Dr. Maher Abbas to discuss and evaluate your condition, click here. If you have previously undergone any operation or tests related to your condition, kindly bring all outside reports for Dr. Maher Abbas to review the day of your consultation.
